医疗器械GMP车间
医疗器械GMP车间解决方案
1、在生产或使用中活性物质、灭活物质的污染(包括热原)对产品产生重要影响的植入性医疗器械,医疗器械GMP车间规划设计,应对工作环境进行控制,对灭活的方法应予验证并保存记录。此类产品的生产和包装应在有规范要求的、可控的环境下进行。
2、对非无菌植入性医疗器械或使用前预期灭菌的医疗器械,如果通过确认的产品清洁、包装过程,能将污染降低并保持一致的控制水平,医疗器械GMP车间施工,应建立一个受控的环境来包含该确认的清洁和包装过程。生产企业可参照YY0033-2000标准或自行验证并确定产品的生产洁净级别。
3、应对受污染或易于污染的产品进行控制。应对受污染或易于污染的产品、工作台面或人员建立搬运、清洁和除污染的文件。
医疗器械GMP车间施工设计参考:
1、《医疗器械生产企业质量管理规范(试行)》,国家食品药品监督管理局(2009年)--2015废止。
2、《体外诊断试剂生产实施细则(试行)》,国家食品药品监督管理局(2007年)--2015废止。
3、《关于实施(医疗器械生产质量管理规范(试行))及其配套文件有关问题的通知》(2011年)--2015废止。
易纯净化补充说明:
前面1~3的2007、2009年医疗器械的规范、细则、标准在2015年停用,医疗器械GMP车间规划,代之2015的医疗器械生产企业质量管理规范及无菌、植入、体外诊断试剂三个附录。
5、《无菌医疗器具生产管理规范》(YY0033-2000)
6、《洁净厂房设计规范》(GB50073-2010)
7、《洁净室施工及验收规范》(GB50591-2010)
……
19、《医疗产品的无菌加工 第1部分:通用要求》(YY/T0567.1-2005)
20、《无菌医疗器械生产与质量管理讲义》,国家药品监督管理局(2000)
23、《无菌医疗器械质量控制与评价》,苏州大学出版社(2012年)
24、《无菌医疗器械生产与洁净厂房的建设》,CMD(2009年)
医疗器械GMP车间等级参考
洁净度级别 | 悬浮粒子最大允许数/立方米 | |||
静态 | 动态(3) | |||
| ≥0.5μm | ≥5.0μm(2) | ≥0.5μm | ≥5.0μm | |
| A级(1) | 3520 | 20 | 3520 | 20 |
| B级 | 3520 | 29 | 352000 | 2900 |
| C级 | 352000 | 2900 | 3520000 | 29000 |
| D级 | 3520000 | 29000 | 不作规定 | 不作规定 |
(1)为确认A级洁净区的级别,每个采样点的采样量不得少于1立方米。A级洁净区空气悬浮粒子的级别为ISO 4.8,以≥5.0μm的悬浮粒子为限度标准。B级洁净区(静态)的空气悬浮粒子的级别为ISO 5,同时包括表中两种粒径的悬浮粒子。对于C级洁净区(静态和动态)而言,空气悬浮粒子的级别分别为ISO 7和ISO 8。对于D级洁净区(静态)空气悬浮粒子的级别为ISO 8。测试方法可参照ISO14644-1。
(2)在确认级别时,应当使用采样管较短的便携式尘埃粒子计数器,避免≥5.0μm悬浮粒子在远程采样系统的长采样管中沉降。在单向流系统中,应当采用等动力学的取样头。
(3)动态测试可在常规操作、培养基模拟灌装过程中进行,证明达到动态的洁净度级别,但培养基模拟灌装试验要求在“最差状况”下进行动态测试。
医疗器械GMP车间配套设备材料
 |  |  |
| 风淋室 | 超净工作台 | 传递窗 |
 |  |  |
| 货淋室 | 洁净取样车 | FFU |
 |  |  |
| 高效送风口 | 净化门窗 | 风幕机 |
 |  |  |
| 快速卷帘门 | 臭氧发生器 | 感应式酒精手消毒器 |
 |  |  |
| 初效过滤袋 | 高效过滤器 | 不锈钢消毒水池 |
 |  |  |
| 风量调节阀 | 可调回风口 | 净化板材 |
 |  |  |
| 净化照明灯具 | 净化铝型材 | 紫外线杀菌灯 |
医疗器械GMP车间设计规范
医疗器械GMP车间工程设计的规范参照:
1、国际标准《ISO/DIS 14644》
2、洁净室厂房设计规范《GB50073-2001》
3、医疗器械包装车间洁净室厂房规范《GMP-97》
4、药品生产质量管理规范《GMP-98》
5、洁净室施工及难收规范《JGJ 71-90》
6、通风与空调工程施工及验收规范《GB 50243-2002》
7、美国联邦标准《FS209E-92》
根据相关规范要求,对无菌医疗器械生产车间、药品生产车间、医学生物学实验室、手术室等都要求建设符合相关标准的洁净室。在洁净室建设或改建时,不能依赖于最终的竣工验收来保证洁净室的质量,必须从设计及设备选型阶段就严格把关,在建设的全过程中对主要关键点严格检查、监督,在实际使用中定期监测才能保证洁净室达到设计指标和使用要求。
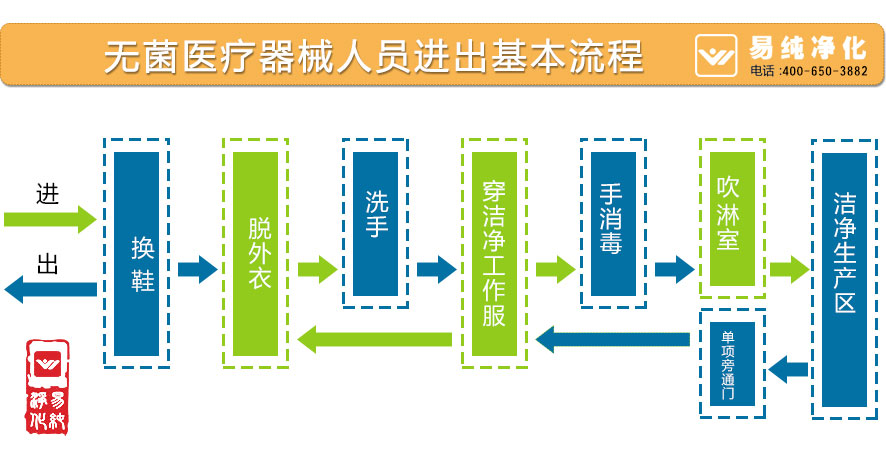
无菌医疗器械是任何标明“无菌”的医疗器械,生产洁净室是保证无菌医疗器械质量的基本条件,控制无菌医疗器械生产过程的环境并规范其生产,防止环境对无菌医疗器械污染,洁净室必须满足规定环境参数的要求来建设和定期监测。

医疗器械净化工程建设中需考虑从以下问题:
1. 医疗器械包装车间洁净室工程所需要的净化材料;
2. 医疗器械厂房洁净室及医疗器械包装车间洁净室工程的设计、安装、调试、维护等综合服务;
3. 医疗器械包装车间洁净室工程空调净化部分
温度和相对湿度
无菌医疗器械在无特殊规定时,通常要求温度在法规标准检测Standard and Testing18~28 C,湿度在 45%~65%,企业一般都可以控制在要求内。如在动态监测中发现达不到要求,可能是室内有产热大的仪器设备。
风量、换气次数、静压差
在洁净室体积确定的情况下,换气次数由该室的送风量决定,而静压差取决于房间的送风量与回风量、排风量的差值。系统总送风量、新风量、总排风量和对外压差可以通过调整风机频率转速或总阀门开启度来实现,各房间的风量和压力则可通过调整分支管路阀门开度来实现。

悬浮粒子、浮游菌、沉降菌
测试条件如不能满足规定的环境参数 ( 温湿度、风速、换气次数、静压差在规定范围之内 ) 要求,关键项目悬浮粒子、浮游菌或沉降菌的测试结果应视为无效。由于温度、相对湿度、风速、换气次数、静压差共同构成了洁净室的微气候,是洁净室维护正常与否的重要指征,可将关键工序关键项目测试修订为关键工序全性能测试。只有这样,才能全面、系统监测生产洁净室,为确保洁净室性能监测的数据科学性、准确性,测试部门在进行关键项目悬浮粒子、微生物测试时,应同时进行温度、相对湿度、换气次数、静压差等前提条件的测试。
温度
洁净室夏季室温超过设计范围的原因,多是由于开始确定的各洁净室的空调送风量即换气次数时只注重满足洁净度指标,忽视了对各洁净室热平衡的校核计算。因此在生产洁净室的设计及运行过程中,必须对洁净室的空调送风参数进行实时修正,保证各个季节生产洁净室的温度都维持 18~28 C。温度和相对湿度主要影响产品生产工艺及细菌的繁殖条件,还能引发由生产操作人员舒适度对产品质量的影响。

送风量、换气次数
医疗器械净化工程-无菌洁净室工程设计阶段对送风量的确定,首先要满足相应洁净度级别的换气次数要求,同时还要通过热、湿负荷校核来进一步确定风量,在此基础上对高效过滤器进行选用。过滤器的处理风量应小于或等于额定风量,设置在同一洁净区内的高效 ( 亚高效、超高效 ) 空气过滤器的阻力、效率宜接近。
医疗器械GMP车间管控总要求:
(1)表面平滑;
(2)表面有耐磨性;
(3)良好的热绝缘性;
(4)不易产生静电;
(5)不吸湿,不透湿;
(6)吸声性好;
(7)容易加工;
(8)表面不易附着灰尘;
(9)容易除去附着的灰尘;
为什么选择无锡易纯净化?






.gif")
.gif")
.gif")
.gif")
.gif")
.gif")
.gif")
.gif")









部分医疗器械净化车间案例
| 建设单位名称 | 工程概况 | 面积 | 净化等级 |
| 解放军第102(常州)医院 | ICU病房 | 800m2 | 10万级 |
| 燕郊京东国济医院 | 无菌手术室 | 230m2 | 1万至10万级 |
| 郑州市骨科医院 | ICU病房 | 600m2 | 10万级 |
| 玛利亚中西医结合医院 | 无菌手术室 | 180m2 | 10万级 |
| 西安惠普生物科技有限公司 | GMP车间 | 800m2 | 10万至30万级 |
| 苏州施莱医疗器械有限公司 | GMP车间 | 900m2 | 10万至30万级 |
| 哈尔滨市眼科医院 | ICU病房 | 620m2 | 10万级 |
| 三德医疗器械(南京)有限公司 | GMP车间 | 500m2 | 10万至30万级 |
| 上海市静安区老年医院 | ICU病房 | 530m2 | 10万级 |
| 重庆长城医院 | ICU病房 | 610m2 | 10万级 |
| 康达医疗器械集团股份有限公司 | GMP车间 | 920m2 | 10万至30万级 |
| 乌鲁木齐市一零四团医院 | 无菌手术室 | 310m2 | 1万至十万级 |
| 济南格利特科技有限公司 | GMP车间 | 670m2 | 10万至30万级 |
| 大通县第一人民医院 | 无菌手术室 | 260m2 | 1万至十万级 |
| 都江堰市人民医院 | 无菌手术室 | 208m2 | 1万至十万级 |
| 南京市鼓楼医院北院 | ICU病房 | 2200m2 | 10万级 |
| 连云港市第二人民医院东院区 | ICU病房 | 1060m2 | 10万级 |
产品质量对比,选择产品一目了然!















电子无尘净化车间案例




咨询热线:135 0619 3709 拨打